Maximizing CPU utilization
As was noticed in previous benchmarkings of the entire workflow, in the HaplotypeCaller phase CPU utilization reached a plateau at approx. 60 - 70 % of all available resources. This can be explained by the fact, that each individual HaplotypeCaller process uses approx. 135 % of the CPU (i.e. 1.35 cores or threads are used). In the Snakefile on the other side only integer values for thread usage are possible. Finally, even though only 1.35 threads are used, 2 threads have to be reserved.
Basically to allow for an overall better CPU utilization of the entire workflow, CPU utilization of HaplotypeCaller has to be shifted to full integer values. To do so, the effects of different settings for the --native-pair-hmm-threads option (1, 2, 4, 6, 8 and 12 threads) were analyzed.
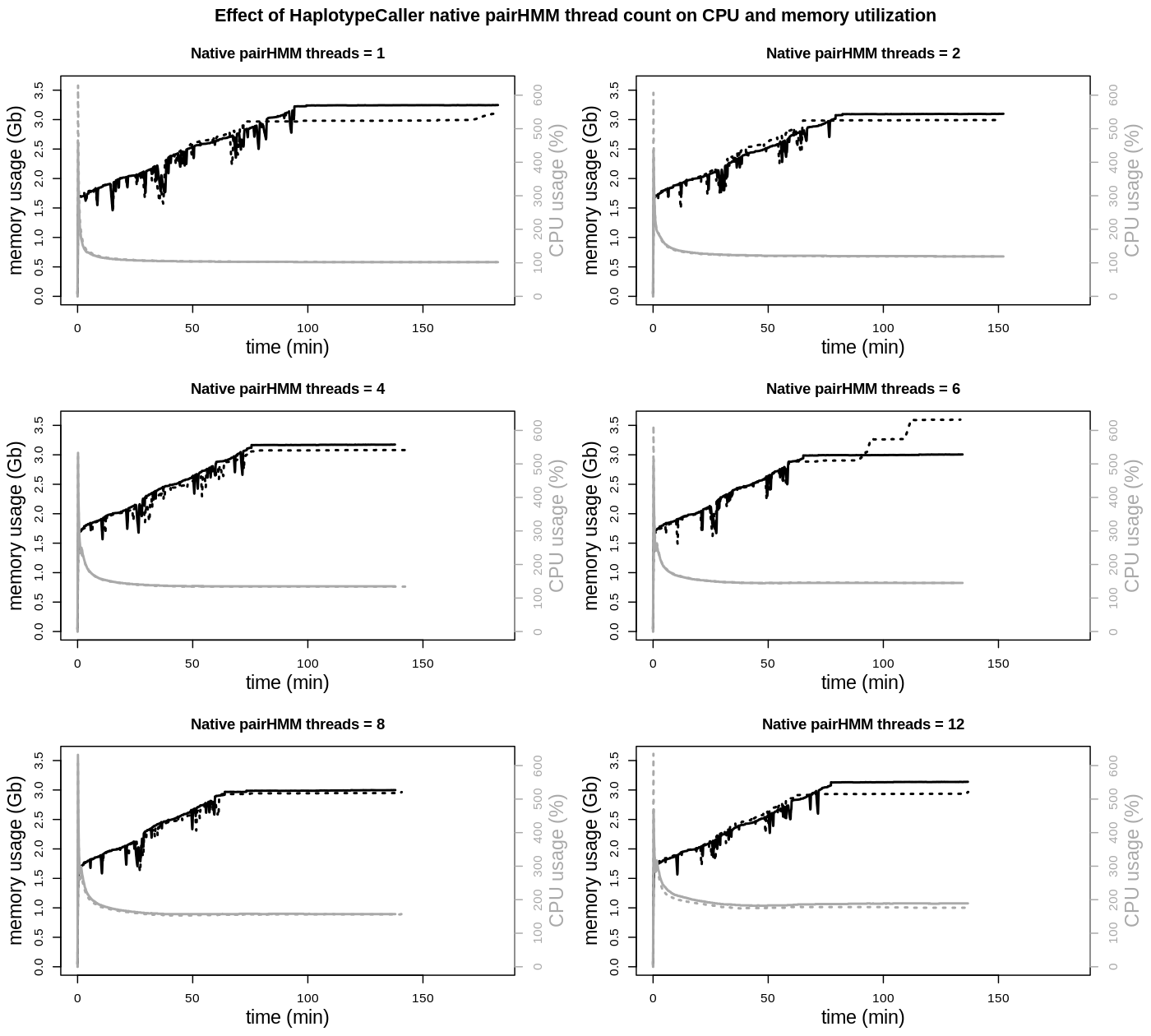
The impact of the number of native pairHMM threads is seen in the total runtimes and also CPU utilization. Total runtimes are declining from 1 to 4 threads, without a noticeable further improvement with higher thread counts. CPU utilization, apart from the always given initial peak load, is continuously rising, as can also be seen from the average CPU utilization:
native pairHMM threads |
average CPU usage (%) |
1 |
106 |
2 |
124 |
4 |
140 |
6 |
152 |
8 |
164 |
12 |
188 |
A higher CPU utilization, close to two threads, is achieved when using 12 native pairHMM threads. But as runtimes are not improved over using 4 threads, there is no benefit in increasing the native pairHMM thread count. On the other side when using only 1 native pairHMM thread, CPU usage is nearly reduced to a single thread. Considering the advantage of being able to run twice the amount of HaplotypeCaller processes in parallel, the slightly increased runtime of the individual process should be negligible.
To test the above hypothesis, the entire workflow was benchmarked again with optimized settings (see previous workflow benchmarking). In two identical data evaluations only the number of native pairHMM threads and the number of Snakemake threads provided to the HaplotypeCaller were altered. Further settings were as follows:
6 intervals per sequencing data set with 6 data sets, resulting in a total of 36 intervals to call
a maximum of 38 Snakemake threads (
--cores 38)-XX:ParallelGCThreads=2for SortSam, MarkDuplicates, HaplotypeCaller and ComineGVCFs-Xmx10Gfor SortSam-Xmx2Gfor MarkDuplicates, HaplotypeCaller and CombineGVCFs
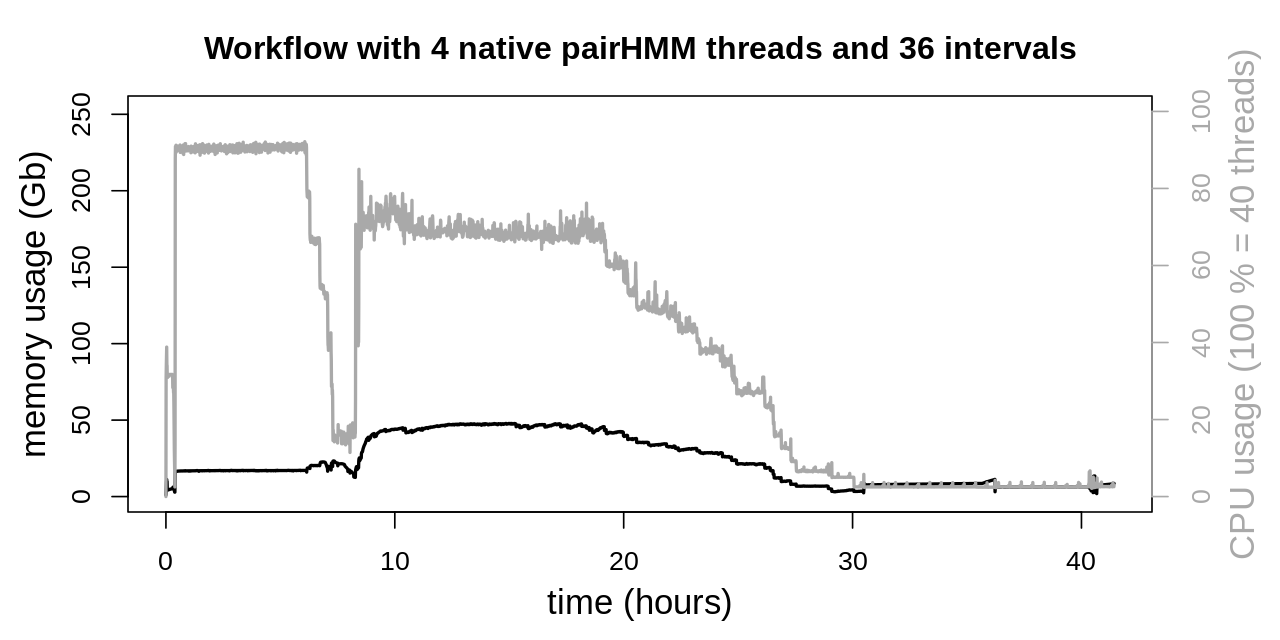
The initial settings reserved 2 threads for every HaplotypeCaller. With a maximum of 38 parallel Snakemake jobs no more than 19 parallel HaplotypeCaller were possible. With this setup again a plateau was reached at approx. 70 % total CPU utilization.
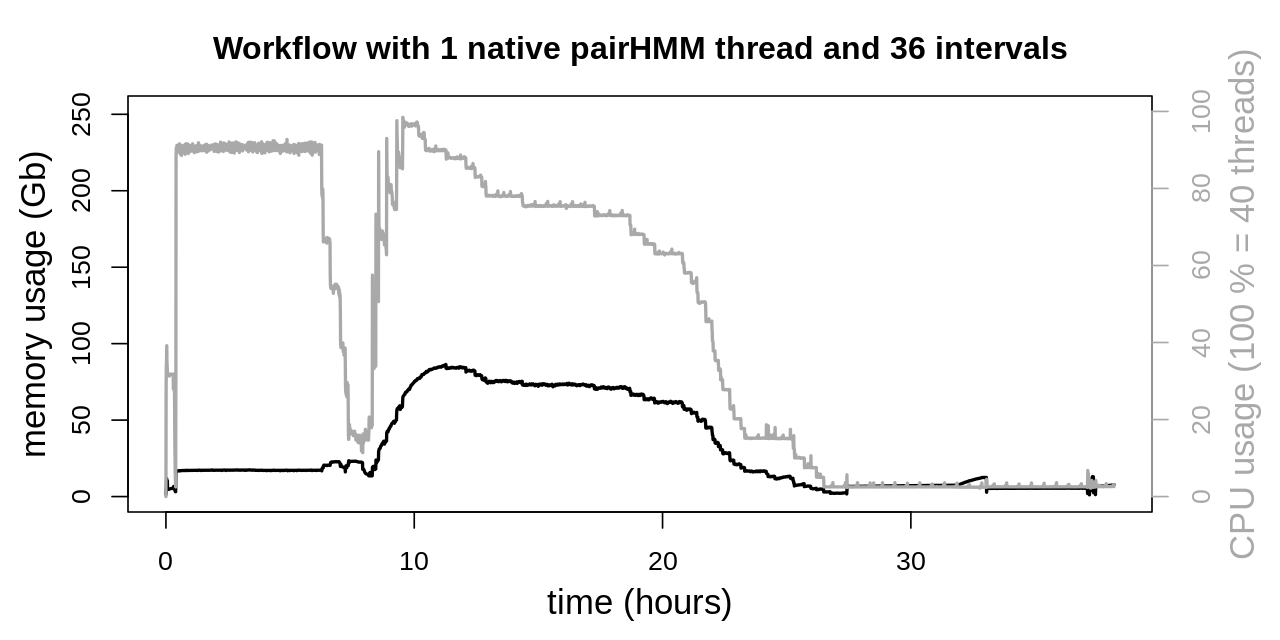
With the reduced CPU usage of a single native pairHMM thread and up to 38 parallel Snakemake jobs, all 36 intervals could be evaluated in parallel by the HaplotypeCaller. As not all intervals are of the same length, short ones are finished before larger ones. This results in a peak load above 90 % total CPU utilization and progressively declining CPU load as more and more HaplotypeCallers are finishing on their respective interval.
When comparing both data evaluations and also previous benchmarkings of the entire workflow, a reduction in the total runtime is observed. When reducing the CPU load of a single HaplotypeCaller but increasing the number of parallel jobs, the entire workflow was finished in 38 h 11 min. On the other side the workflow without reducing performance of the individual HaplotypeCaller was finished in 41 h 26 min. This clearly shows the overall runtime advantage of the higher parallelization, despite the individual HaplotypeCaller being less performant.
The above procedure is especially interesting when hardware resources are limited and a maximum degree of system utilization is desirable. On the other hand, if hardware resources are not a limiting factor, for instance with a large cluster that would idle anyways, not limiting the individual HaplotypeCaller would result in a slightly reduced runtime. In such circumstances the default of 4 native pairHMM threads is the optimum. For general usage OVarFlow includes the default setting of four native pairHMM threads. Configuration is enabled through the config.yaml file.